|
|
|
|
|
QB期刊 | stIHC:一种识别空间基因共表达的新方法 |
|
|
论文标题:Spatial transcriptomics iterative hierarchical clustering (stIHC): A novel method for identifying spatial gene co-expression modules
期刊:Quantitative Biology
作者:Catherine Higgins, Jingyi Jessica Li, Michelle Carey
发表时间:28 Aug 2025
DOI:10.1002/qub2.70011
微信链接:点击此处阅读微信文章
空间转录组学(ST)技术的最新进展使研究人员能够在保留组织内空间信息的同时,测量数百至数千个基因的RNA表达水平,为空间基因表达模式、组织结构和基因功能提供了关键见解。然而,现有的将空间可变基因(SVGs)聚类为共表达模块的方法往往无法检测罕见或独特的空间表达模式。
近日,美国加州大学洛杉矶分校的Jingyi Jessica Li团队在Quantitative Biology期刊上发表了题为“Spatial transcriptomics iterative hierarchical clustering(stIHC): A novel method for identifying spatial gene co‐expression modules”的方法。作者提出的空间转录组学迭代层次聚类(stIHC),是一种将SVGs聚类为共表达模块的新方法,代表具有共享空间表达模式的基因组。通过三项模拟实验以及对10x Visium、10x Xenium和Spatial Transcriptomics等技术的ST数据集的应用,stIHC优于SPARK、SPARK-X、MERINGUE和SpatialDE等流行SVG检测方法所使用的聚类方法。基因GO富集分析证实,每个模块内的基因具有一致的生物学功能,支持空间共表达的功能相关性。stIHC在不同基因数量和空间分辨率的技术中均表现出稳健性,为解码基因表达的空间组织和复杂组织的功能结构提供了强有力的工具。

全文概要
空间转录组学技术能同时测量基因表达和保留组织空间信息,有助于揭示空间基因表达模式。但现有方法在将空间可变基因聚类为共表达模块时,往往难以检测到罕见或独特的空间表达模式。本文提出的stIHC 方法是一种新颖的两步聚类法,专门用于将 SVGs 聚类为共表达模块(即具有相似空间表达模式的基因组)。
stIHC方法介绍
stIHC方法分为两步:
第一步:基因表达建模。使用广义惩罚回归框架(generalized penalized regression framework)对二维组织切片中的基因表达进行建模。该方法首先通过Delaunay三角剖分将感兴趣的组织切片划分为更小的区域,这是一种处理非规则几何形状(如不规则组织形状)的高效算法。然后在三角形上定义局部支撑的多项式函数作为基函数,采用惩罚迭代加权最小二乘法估计系数。这一步骤至关重要,因为原始基因表达数据通常包含大量噪声,而通过建模可以将基因在空间位点的表达水平视为光滑连续函数而非离散值,从而有效降低噪声。
第二步:基因聚类。使用功能型迭代层次聚类算法(funIHC)基于估计的模型系数对SVGs进行聚类。该方法采用Spearman相关系数构建距离度量,通过"聚类-合并-剪枝"的迭代策略处理不平衡簇,能够识别仅含单个基因的独特空间表达模式,并自动确定最优簇数而无需用户预设参数。
stIHC方法评估
作者通过三项模拟实验评估了stIHC 在空间转录组数据上的聚类性能,并将其与四种 SVG 检测方法中所使用的聚类方法(SpatialDE、SPARK、SPARK‐X和 MERINGUE)进行比较。结果发现,在平衡簇的理想场景下,所有方法表现相当;但在不平衡簇的模拟中,stIHC是唯一正确识别所有模块的方法,而SPARK、SPARK-X和MERINGUE都遗漏了最小的两基因簇,SpatialDE则完全错误地识别了空间模式(图1)。
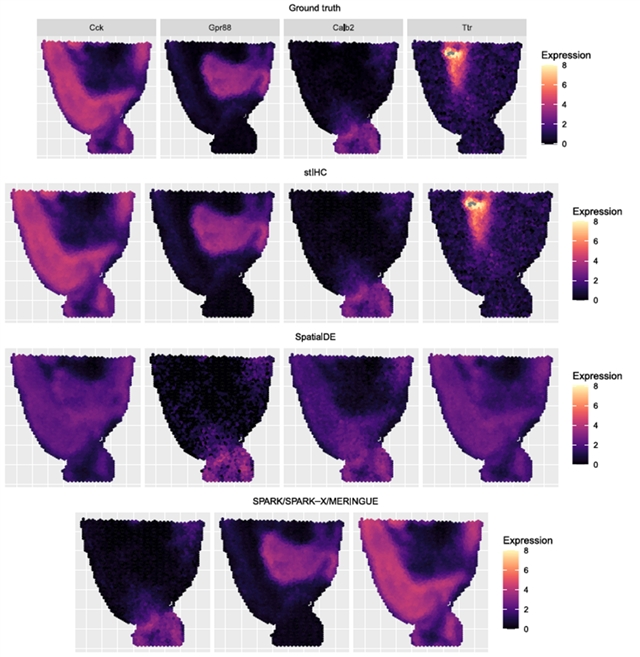
图1. stIHC与其他四种聚类方法(SpatialDE、SPARK、SPARK‐X和 MERINGUE)在不平衡簇的模拟数据中的比较
在稀疏分辨率的不平衡场景下,stIHC依然保持最优表现,是唯一能够保留最小模块空间模式的方法(图2)。此外,stIHC具有完全可重复性,不引入随机初始化带来的变异,计算效率也较为合理。
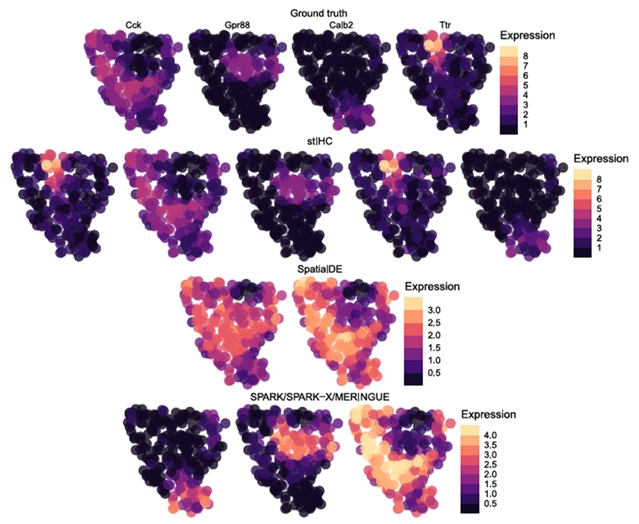
图2. stIHC与其他四种聚类方法(SpatialDE、SPARK、SPARK‐X和 MERINGUE)在稀疏分辨率的不平衡场景中的比较
在三个真实数据集的应用中,stIHC展现出强大的生物学发现能力。在10x Visium(图3A)和10x Xenium(图4)两种不同技术平台的小鼠脑数据中,该方法一致识别出对应于海马、丘脑和下丘脑的基因模块,GO富集分析显示这些模块分别与记忆学习、感觉信号传递和激素调节等功能相关,与已知的脑功能解剖结构高度吻合。在人肺癌数据中(图5),stIHC识别出两个模块,一个对应正常肺功能和免疫反应,另一个与肿瘤生长相关的肽激素加工、止血和组织稳态等通路相关,显示出潜在的临床意义。在小鼠嗅球数据的应用中,stIHC展示了与不同SVG检测方法(MERINGUE和SpatialDE)的兼容性,能够自动确定最优簇数并识别与已知解剖层对应的基因模块。
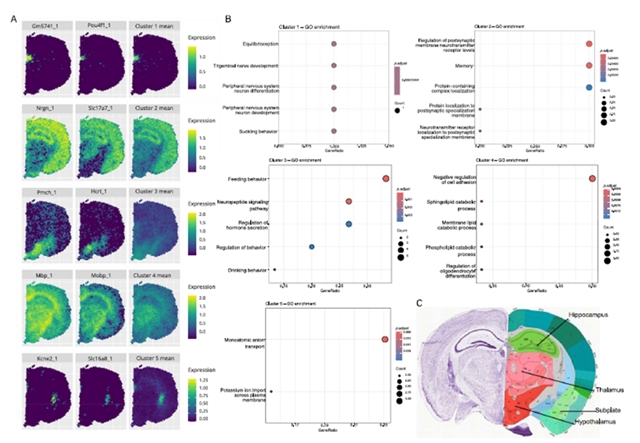
图3. stIHC在10x Visium平台中小鼠脑数据中的表现
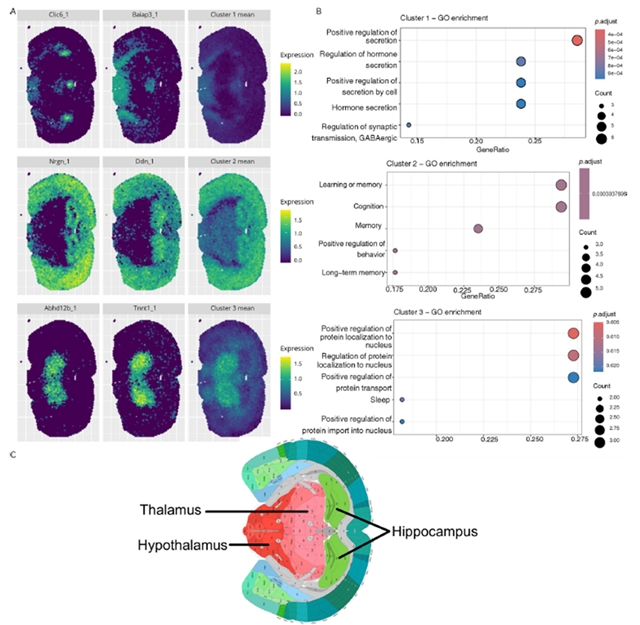
图4. stIHC在10x Xenium平台中小鼠脑数据中的表现
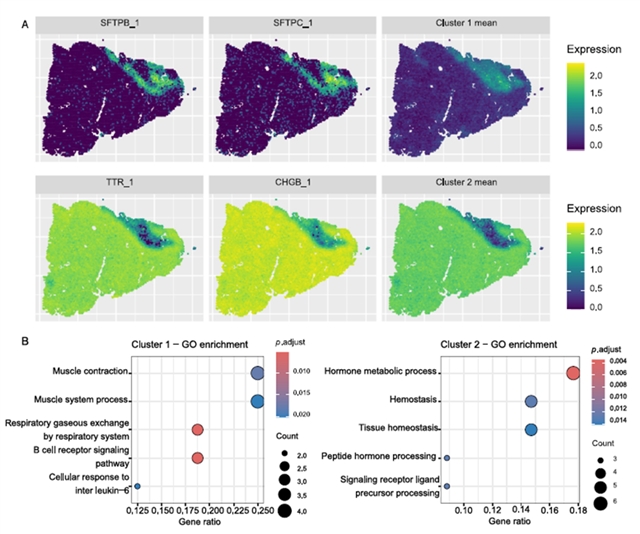
图5. stIHC在10x Visium平台中肺癌数据中的表现
总结
stIHC的核心优势在于其专门设计用于处理不平衡基因共表达模块。传统方法在平衡数据集上表现良好,但在不平衡场景下倾向于将小簇合并到大簇中,导致罕见但生物学重要的空间模式被遗漏。通过结合广义惩罚回归的噪声降低能力和funIHC的不平衡簇处理能力,stIHC的优势如下:(1)识别稀有空间模式:即使模块仅含单个基因也能正确识别,这对检测稀有细胞类型或病理特异性表达模式至关重要;(2)保证结果可重复性:无随机初始化或随机过程带来的变异,确保特定数据集上结果完全可重复;(3)实现参数精简:自动确定最优簇数,无需用户预设参数,降低使用门槛;(4)保持跨平台稳健性:在不同ST技术平台(测序-based和成像-based)上均表现一致;(5)提供生物学可解释性:识别的模块与已知解剖结构和功能通路高度吻合。
综上所述,stIHC为解码复杂组织的空间基因表达组织和功能结构提供了一个数据驱动、计算高效且生物学可解释的强有力工具。其R实现已在GitHub开源发布(CatherineH1/stIHC),所有模拟数据和真实数据集也已公开,便于其他研究者复现和扩展该方法的应用。
QB期刊介绍
Quantitative Biology (QB)期刊是由清华大学、北京大学、高等教育出版社联合创办的全英文学术期刊。由高等教育出版社和Wiley双平台出版和发行。QB主要刊登生物信息学、计算生物学、系统生物学、理论生物学和合成生物学的最新研究成果和前沿进展,并为生命科学与计算机、数学、物理等交叉研究领域打造一个学术水平高、可读性强、具有全球影响力的交叉学科期刊品牌。
QB期刊目前已被ESCI, PMC, Scopus, DOAJ, CSCD等国内外重要数据库收录。
在线浏览
https://journal.hep.com.cn/qb/EN
https://wileyonlinelibrary.com/journal/qb
投稿
https://mc.manuscriptcentral.com/qb
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。






