3月24日,中山大学肿瘤防治中心教授张力、方文峰,副研究员李婧团队成功揭示表皮生长因子受体(EGFR)融合突变的致癌新机制。相关成果发表于《癌症发现》(Cancer Discovery)。
基因组不稳定性是恶性肿瘤的核心特征之一,基因融合突变作为其重要表现形式,不仅是多种实体瘤发生发展的关键驱动事件,也是导致治疗耐药的重要因素。在肺癌精准治疗领域,以受体酪氨酸激酶(RTK)为核心的融合基因被视为“钻石靶点”。长期以来,受“激酶中心论”影响,学术界普遍认为融合蛋白的致癌驱动力主要源于激酶结构域的异常激活,临床治疗高度依赖酪氨酸激酶抑制剂(如EGFR-TKI)的单药干预。然而,越来越多临床证据表明,部分融合驱动的肿瘤对激酶抑制剂响应有限,甚至出现原发耐药,提示融合基因的致癌机制中可能存在被忽视的非激酶依赖路径。
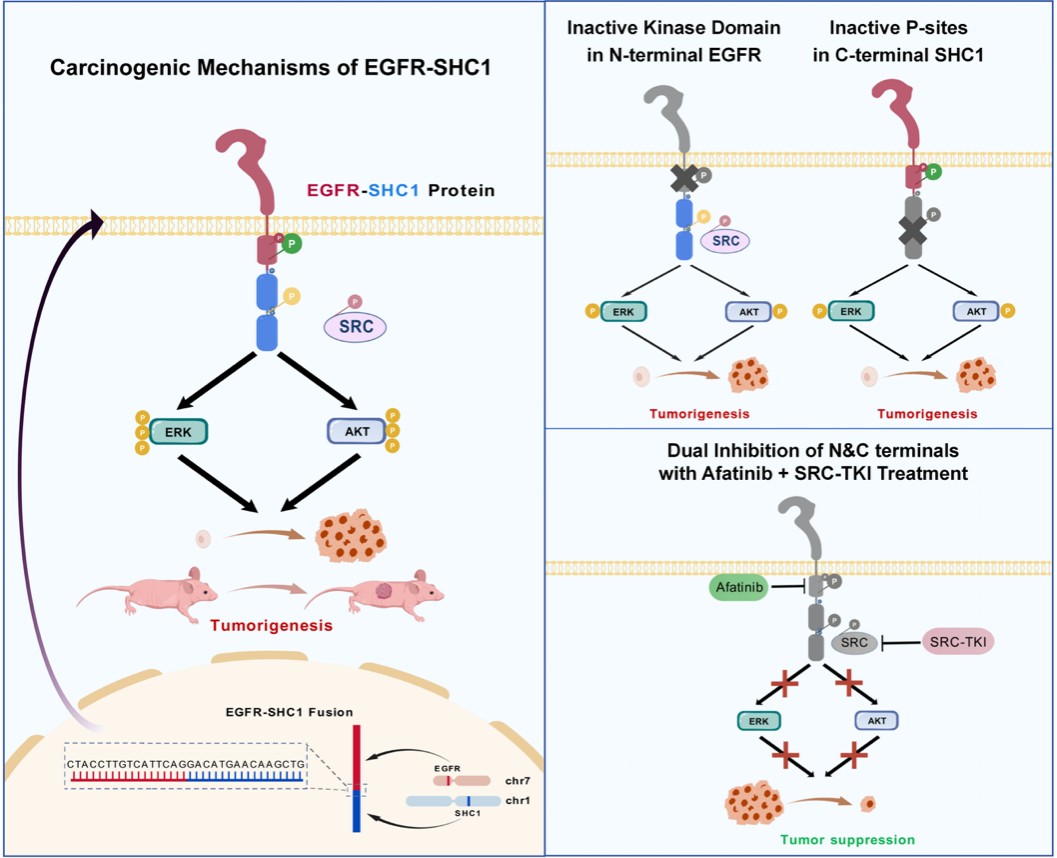 耐药机制模式图。研究团队供图
耐药机制模式图。研究团队供图
研究团队通过深入分析真实世界中的EGFR-SHC1融合病例,提出RTK融合基因的一种全新“双激活”致癌模式:融合蛋白通过N端激酶活性与C端融合伴侣蛋白的磷酸化位点协同作用,共同诱导细胞转化。这一发现挑战了长期主导该领域的“激酶中心论”,表明融合伴侣并非被动配角,而是主动驱动致癌信号重编程的关键因素。基于该理论模型,团队提出的“双靶点拦截”联合治疗策略,为破解RTK融合相关内在耐药提供了新的理论框架与临床解决方案。
研究团队在肺腺癌患者中鉴定出一类由7号染色体EGFR与1号染色体SHC1易位形成的融合基因,该基因保留了EGFR的激酶域与SHC1的接头功能域。功能实验证实,EGFR-SHC1呈现“双激活”模式:一方面通过N端EGFR激酶域触发信号传导,另一方面通过C端SHC1的磷酸化位点直接驱动下游ERK/AKT通路。机制研究表明,即使在EGFR激酶活性被TKI有效抑制的情况下,SRC激酶仍能通过磷酸化C端SHC1建立旁路信号,维持肿瘤细胞存活。这一机制解释了该亚型对EGFR-TKI单药治疗产生内在耐药的分子基础。
基于上述发现,团队提出了“EGFR-TKI(阿法替尼)联合SRC抑制剂(达沙替尼)”的双靶点拦截策略。体外实验及临床治疗结果均显示显著的协同抑制效果:在一例对TKI单药原发耐药的患者中,该联合方案成功诱导了明显的肿瘤退缩。这项“从实验室到病床”的转化研究,为此类难治性融合亚型提供了可行的临床决策依据。
该研究拓展了RTK融合突变的致癌模型,证实融合伴侣并非简单“挂件”,而是能够建立平行、激酶非依赖信号轴的核心驱动力。这一发现揭示了“激酶-融合伴侣”协同致癌的新机制,为理解RTK融合的异质性提供了新的理论框架。在临床解读基因检测报告时,需突破“激酶中心论”的传统认知,系统评估融合伴侣的功能属性,以制定更精准的治疗方案。
相关论文信息:https://doi.org/10.1158/2159-8290.CD-25-1936
版权声明:凡本网注明“来源:中国科学报、科学网、科学新闻杂志”的所有作品,网站转载,请在正文上方注明来源和作者,且不得对内容作实质性改动;微信公众号、头条号等新媒体平台,转载请联系授权。邮箱:shouquan@stimes.cn。






