|
|
|
|
|
当沙门氏菌越来越“难对付”,大模型与量子计算如何识别它的耐药倾向? |
|
|
四川大学章乐教授团队联合相关研究人员在中国工程院院刊Engineering发表题为“Developing a Predictive Platform for Salmonella Antimicrobial Resistance Based on a Large Language Model and Quantum Computing”的研究性文章。该研究围绕沙门氏菌抗菌药物耐药性预测问题,结合泛基因组分析、大语言模型与基于量子计算的数据增强方法,构建了一个耐药性预测框架与在线平台,旨在从复杂基因组数据中提取与耐药相关的关键信号,为病原体耐药风险研判、食品安全监测和公共卫生预警提供参考。
从“测出来”到“看出来”:沙门氏菌耐药性为什么越来越难识别?
沙门氏菌耐药性的形成,并不是由单一因素决定的,而往往与多个耐药相关基因及其复杂相互作用密切相关。随着基因组测序技术的发展,研究者能够获取越来越丰富的病原体遗传信息,但数据更多,并不意味着识别更容易。相反,真正的难点在于:如何从海量而复杂的基因组数据中,找出与耐药表型真正相关的关键信号。与此同时,耐药性预测不仅依赖模型本身的学习能力,也高度依赖输入特征的质量与有效性。如果关键耐药信息被大量冗余特征和噪声信号所掩盖,模型即使能够完成训练,也难以准确识别耐药规律。因此,在开展沙门氏菌耐药性预测之前,首先需要解决的,就是复杂基因组背景下关键耐药相关特征的识别与筛选问题。
大语言模型进入耐药性预测:它为什么不仅是“会生成文本”的工具?
在完成关键耐药相关特征筛选之后,接下来的问题是:怎样建立一个能够更有效识别耐药规律的预测模型。近年来,人工智能技术在生物医学领域不断拓展应用,其中,大语言模型在复杂模式识别与表征学习方面提供了新的思路,也为病原体耐药性分析带来了新的方法选择。与传统机器学习方法相比,这类模型更关注基因组特征与耐药表型之间潜在而复杂的关联关系。基于这一思路,研究进一步构建了面向沙门氏菌耐药性预测的大语言模型方法,并将筛选得到的关键特征输入模型,以识别基因组特征与耐药表型之间的对应关系。如图1所示,该研究以泛基因组分析为基础,结合特征筛选、模型训练与平台构建,形成了一个面向沙门氏菌耐药性预测的整体研究流程。由此,大语言模型在这里并不是用于常规文本生成,而是通过将基因组特征文本化并进行任务适配,被引入到沙门氏菌耐药性预测这一具体生物医学场景之中。
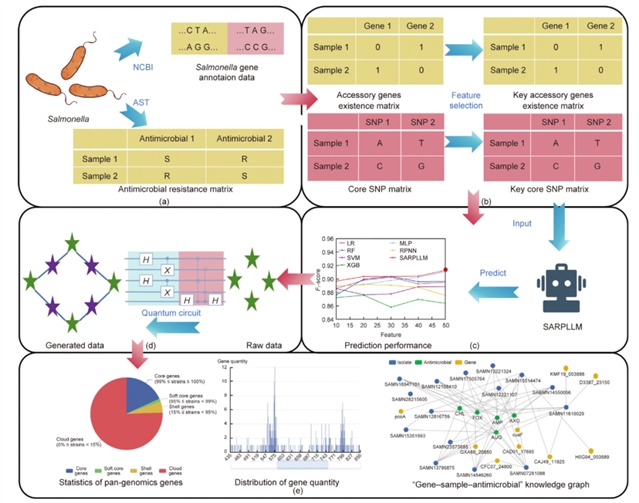
图1.从基因筛选到在线预测平台的总体研究流程
基于量子计算的数据增强方法为什么也被引入进来:从模型预测到在线应用还差哪一步?
在耐药性预测任务中,模型性能的提升不仅取决于特征筛选和算法设计,也与数据处理方式密切相关。对于复杂生物数据而言,样本分布不均衡、特征空间高维以及计算效率受限,都会对预测结果的稳定性和推广能力产生影响。因此,在提高模型预测能力之外,如何进一步优化数据增强过程、提升计算效率,并推动模型从研究走向实际应用,同样是这一研究需要解决的问题。围绕这一目标,本文研究进一步引入了基于量子计算的数据增强方法,并在此基础上构建了面向用户的在线沙门氏菌耐药性预测平台。一方面,量子启发算法被用于优化样本处理过程,用于优化样本距离计算过程,以提升在样本不均衡和高维数据条件下的数据处理效率;另一方面,研究者将预测模型、分析结果与知识图谱展示整合到平台之中,使沙门氏菌耐药性预测不再停留于算法层面,而是进一步具备了在线分析、结果呈现与信息共享的应用基础。如图2所示,该平台围绕耐药性预测任务,集成了模型分析、结果展示与信息服务等功能。

图2.沙门氏菌耐药性预测在线平台
从模型构建到实际应用:沙门氏菌耐药性预测还面临哪些问题?
尽管基于人工智能的沙门氏菌耐药性预测正在不断推进,但从方法设计走向稳定应用,仍有一些问题需要继续处理。首先,病原体耐药性的形成机制本身较为复杂,不同菌株、不同药物类别以及不同数据来源之间,往往都存在明显差异,这会直接影响模型在更广泛场景中的适用性。其次,预测结果不仅取决于模型结构,也与训练数据的规模、质量和代表性密切相关。如果数据来源有限,或者不同类型样本之间分布不均,模型在实际使用中的稳定性就会受到影响。与此同时,从研究走向应用,还需要进一步兼顾计算效率、结果可解释性以及平台可用性等方面的问题。也就是说,沙门氏菌耐药性预测若要进一步服务于食品安全监测、公共卫生预警和病原体风险评估,还需要在预测稳定性、数据代表性、计算效率和平台可用性等方面持续完善,不仅需要“能预测”,还需要“预测得稳、用得起来”。
结果与讨论
本文关注的并不是单一算法性能的提升,而是围绕沙门氏菌耐药性预测这一问题,尝试建立一套从关键特征识别、模型构建到在线应用的平台化方法。其核心思路在于,将泛基因组分析、大语言模型与量子计算结合起来,从复杂基因组数据中提取耐药相关信号,并进一步形成可用于在线分析与结果展示的预测流程。这样的工作所对应的,不只是模型层面的耐药性判断能力提升,也是在探索如何将计算方法更系统地引入病原体耐药风险研判之中。
从更广的意义上看,沙门氏菌耐药性预测的价值,不仅体现在对单个菌株耐药倾向的识别上,也体现在其对食品安全监测、公共卫生预警和病原体风险评估的支撑作用上。对于这类问题而言,真正重要的并不只是“得到一个预测结果”,而是如何在复杂数据背景下,更早识别耐药风险、提高分析效率,并为后续研判提供更具参考价值的信息。这也说明,面向病原体耐药性的预测研究,正在从单纯的方法探索,逐步走向与实际应用场景相结合的分析工具建设。(来源:EngineeringJournals微信公众号)
相关论文信息:https://www.sciencedirect.com/science/article/pii/S209580992500030X?via%3Dihub
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。