|
|
|
|
|
【前沿速递】一种超越常规认知之外的三硫桥联双核吡啶二亚胺铱配合物的形成与可逆断裂 | MDPI Inorganics |
|
|
论文标题:Formation and Reversible Cleavage of an Unusual Trisulfide-Bridged Binuclear Pyridine Diimine Iridium Complex
论文链接:https://doi.org/10.3390/inorganics14010011
期刊名:Inorganics
期刊主页:https://www.mdpi.com/journal/inorganics
一、引言
铱的吡啶二亚胺(PDI)配合物是构建高反应性的Ir–氮烯(Ir–nitrido)物种的基础。这类物种具有显著的多重键特征,能够活化H–H、C–H、Si–H,甚至C–C键。来自德国汉堡大学的Burger教授团队此前制得了一个由PDI配体支持的端位Ir–SH配合物,并指出其中Ir–S键带有明显的多重键特征。基于这一结果,他们把该Ir–SH物种视为进一步获得端位Ir–S物种的潜在前体,并尝试借助氢原子转移(HAA)来实现这一目标。这项工作的主要意义在于:端位金属–硫配合物在第5、6族过渡金属中已有较多报道,但在后过渡金属中基本仍属未知,因此这项工作本身就是在探索后过渡金属端位M–S体系能否被真正捕捉的证据。
二、研究过程与结果
研究团队把2,4,6-三叔丁基苯氧自由基(TTBP)加入硫醇盐配合物1和2。反应后溶液颜色由深紫色变成深蓝/紫色;其中配合物3被分离出来,分离收率为 1%。从核磁上看,起始物里能清楚看到的硫醇氢特征峰在产物中消失,同时出现对应酚产物的新信号。这一组变化可被直接解读为:HAA确实发生在Ir-SH位点上。
作者最开始预想,HAA之后也许会产生一个双核的二硫桥联产物(图1所示)。
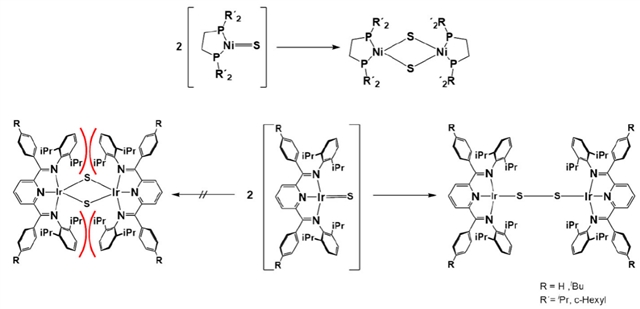
图1. 双核镍(上)与铱(下)二硫桥配合物的形成(红色弧线标示了金属–硫结构单元周围位阻显著拥挤的区域)
但单晶X-ray最终给出的结构并不是Ir2S2,而是一个三硫桥物种,也就是PDI-Ir-S3-Ir-PDI(如图2所示)。
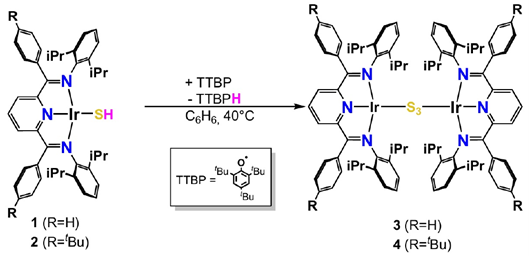
图2. 硫醇盐配合物与 TTBP 自由基的反应示意图(反应式未严格配平)
研究团队发现用于支撑Ir2S3结构的证据链还包括:二维核磁DOSY NMR显示产物尺寸大于单核前体;单晶结构可以直接观察到三硫桥的存在;MALDI与LIFDI都给出与Ir2S3对应的分子离子峰;而用于质子计量的外标实验表明其质子数更接近S3桥联模型,而不是S2桥联模型。除此以外,研究团队还给出了关键的结构信息:配合物3呈C2对称;两个Ir中心处于同一平面;Ir–S键长为2.2263Å,S–S键长为2.0850Å,S–S–S键角为101.47°。研究团队还指出,这个Ir–S键比前体1和2中的Ir–S键更短(图3)。
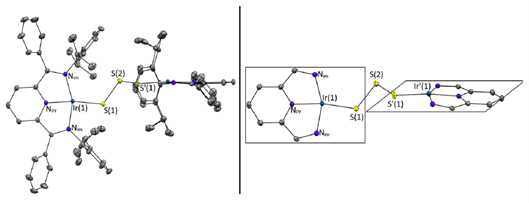
图3. 化合物3的分子结构Ortep图。左:为清晰起见,省略氢原子,热椭球以50%概率水平绘制。右:为进一步简化图示,还省略了芳基和苯基。(碳原子,灰色;氮原子,蓝色;铱原子,青绿色;硫原子,黄色。)
这正是本项工作真正有意思的地方,因为它恰恰“没有得到原本想要的东西”。研究团队本来想借HAA走向一个端位Ir–S单元;但实验上并没有把这个物种直接抓住,反而得到一个异常的三硫桥联双核产物。换句话说,体系自发走向了一个更稳定、也更少见的Ir–S3–Ir产物。
三、Ir–S3–Ir物种形成的机理解释
计算化学结果表明这一步的发生并不困难:研究团队给出的LNO-CCSD(T)//DFT结果为ΔG298 = -12.7 kcal/mol,而过渡态势垒约为18.1 kcal/mol。图4展示了这一步的过渡态,图5则显示生成的Ir-S•中间体自旋密度主要集中在硫原子上。
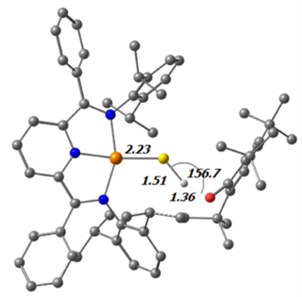
图4. 配合物1与TTBP自由基反应过渡态的计算结构:Ir–S–H + •O–(2,4,6-tBu)Ph → IrS• + H–O–(2,4,6-tBu)Ph。图中标注了部分关键键长(Å)和键角(°)
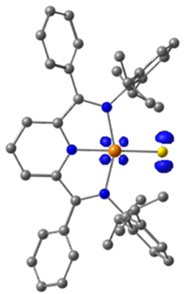
图5. 暂定HAA中间体Ir–S•自由基的自旋密度分布,由DFT计算得到(等值面值 = 0.02 e−/Å3)
研究团队随后提出:这个Ir–S•中间体应当很容易先二聚成Ir–S2–Ir,因为对应的DFT结果显示这一过程几乎无势垒,而且热力学上有利。也就是说,Ir–S2–Ir更像是一个很快形成的早期中间体,而不是最终停留的终点(图6)。
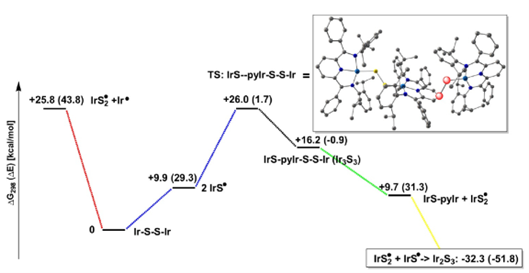
图6. 生成三硫桥结构物种的二聚反应机理自由能变化图(DFT计算水平:PBE-D4/def2-TZVP)
接下来的关键问题是:第三个硫从哪里来?研究团队讨论后推测,直接把额外的Ir–S•加到拥挤的二硫桥核心上并不合理;正如图7所示,空间填充模型表明这一步位阻过大。于是作者转而提出一条由DFT支持的替代路径:Ir–S•从吡啶对位后侧进攻Ir–S2–Ir,先形成加成体,再释放IrS2•,最后导向Ir–S3–Ir产物。
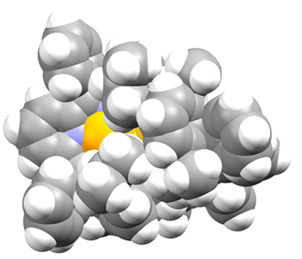
图7. IrS2Ir的空间填充模型
随后研究团队把配合物3在H2气氛下加热。如图8所示,双核物种的特征信号会逐步消失,同时原先属于单核含硫氢根(SH)配体的配合物1的硫醇氢单峰信号又重新出现;最终得到的1H和13C NMR与单核硫醇盐物种一致,这明确表明:热解后又回到了单核物种。
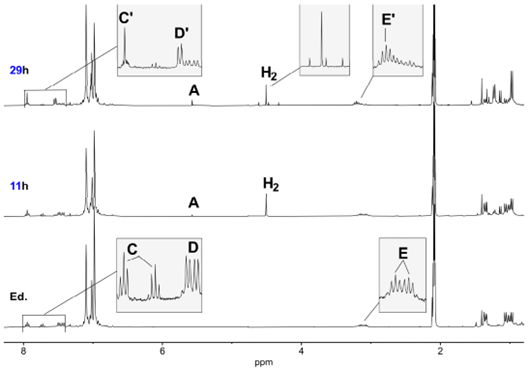
图8. 化合物3在甲苯-d8中、氢气存在下热解反应的1H NMR谱(300 MHz)。底图为配合物3(反应底物)的谱图;中图为加入氢气后在80 °C下反应11 h的谱图;上图为在80 °C下反应29 h后的谱图。A = SH,C/C′ = H-py,D/D′ = H-2,6-ph,E/E′ = H-1-dipp
结合前述机理实验和计算化学结果,研究团队更倾向于把再生的S–H解释为分子内氢转移,而不是溶剂供氢。如图9所示,研究团队给出了讨论的几条可能C–H活化/内扣型路径,核心要点是:配体上的氢,尤其是DIPP上的氢,更可能参与回到金属-硫单元;但同时需要指出的是,研究团队并没有在1H NMR中观察到能直接支持该类抗磁性内扣型中间体中的氢化物信号。
四、总结与讨论
Burger团队在这项工作中考察了硫醇盐配合物与苯氧自由基之间的反应性,目标是理解这类体系如何形成μ-硫桥双核物种。原本按照“最小构型变化”的思路,最有可能得到的应该是二硫桥 Ir–S2–Ir配合物;但实验结果却出人意料:作者最终分离并通过单晶X射线衍射与质谱明确表征到的是一个三硫桥Ir–S3–Ir物种。
进一步的DFT计算表明,这一结果并非偶然。相比于对应的IrS2Ir结构,IrS3Ir骨架在热力学上更稳定,因此体系更倾向于形成μ-三硫桥,而不是直觉上更容易想到的μ-二硫桥结构。也就是说,实验现象与理论计算在这里给出了高度一致的答案。
研究团队随后还继续追踪了这一桥联双核物种的后续反应性。结果发现,无论是在氢气气氛下热解,还是在更高温、无氢气条件下处理,该体系最终都能够回到单核硫醇盐配合物。这说明三硫桥结构虽然可以形成并被稳定捕获,但在一定条件下又能够发生转化,体现出这一硫桥构筑过程具有一定的可逆性与动态平衡特征。
Inorganics 期刊介绍
主编:Duncan H. Gregory, University of Glasgow, UK
期刊范围涵盖固体无机化学、配位化学、生物无机化学、有机金属化学、无机材料化学、理论无机化学、超分子化学和应用无机化学等,着重报道新的和已知无机化合物的合成、热力学、动力学性质、谱学、结构和成键等性能。
|
2024 Impact Factor
|
3.0
|
|
2024 CiteScore
|
4.1
|
|
Time to First Decision
|
14.9 Days
|
|
Acceptance to Publication
|
2.8 Days
|
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。






