|
|
|
|
|
学者开发出基因编辑突变分析新工具SuperDecode |
|
|
近日,《分子植物》(Mol Plant)发表了华南农业大学农学院教授刘耀光/谢先荣团队同合作者的最新研究成果。他们开发了一个基于Sanger测序、二代测序和三代测序技术对各种基因编辑样品的不同类型突变进行高效检测的方法和分析工具箱SuperDecode。目前,SuperDecode提供了Windows、MacOS本地化界面版和Linux命令行版,以及网页版。
 SuperDecode工具箱的分析模块和功能。研究团队供图
SuperDecode工具箱的分析模块和功能。研究团队供图
?
基因组编辑技术已成为基因功能研究和遗传操作的革命性工具,广泛应用在生命科学的多个领域。随着基因编辑技术的发展和应用场景的多元化,所产生的突变类型更加多样,需要的检测样本数也在大幅度的增加,如何更加高效、低成本的对编辑结果进行鉴定已成为亟待解决的问题。目前对基因编辑材料的突变类型鉴定,主要是通过Sanger测序和二代测序技术进行检测。
然而,Sanger测序无法检测嵌合突变、频率低或复杂类型的突变,并且受到通量低的限制,使用Sanger测序对大规模样本进行检测的成本较高。基于二代测序的突变检测技术提高了样本突变检测的通量,低频率突变和复杂突变的检测能力也高于Sanger测序,但是,由于读长短的特点,基因组大片段的插入、删除以及在一定区域内多靶点编辑引起的结构变异,难以使用二代测序技术进行检测。
为满足科研人员在不同应用场景下的检测需求,研究团队开发了一个全面、综合的基因编辑突变分析工具箱SuperDecode,可以实现对利用不同测序平台产生的数据进行高效分析,并且优化了基于PCR构建多样本混合文库的方法。
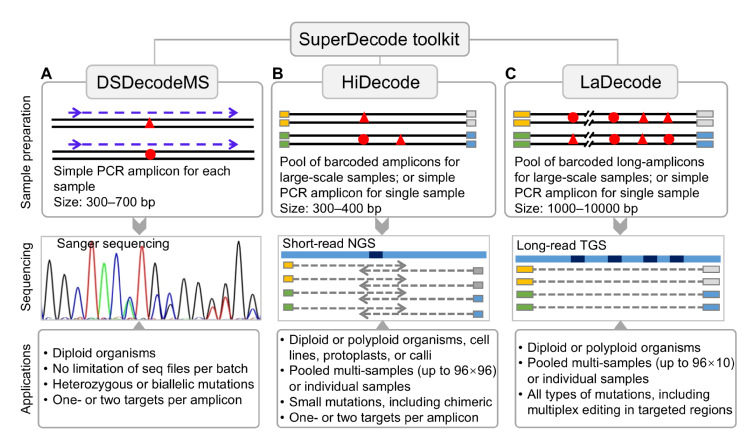
据介绍,SuperDecode包含3个子软件模块:DSDecodeMS、HiDecode以及LaDecode,分别对应Sanger测序、二代测序结果以及三代测序数据的解码分析。其中,DSDecodeMS是研究团队此前开发的网页版工具DSDecode/DSDecodeM的升级本地版,添加了去除Sanger测序两端低质量序列的功能,具有更快的分析速度以及更为友好的使用界面,可通过直接读取靶点扩增子的Sanger测序峰图,分析样本的突变类型。
HiDecode能对添加特定barcode序列的多样本混合文库的二代测序文件进行解码,实现多种类型样本的高通量突变分析,包括二倍体、多倍体、细胞系等。同时,HiDecode中提供了96 × n种特异性barcode序列,理论上可一次性检测多达9,216(96 × 96)个样本的突变类型。LaDecode则是基于单分子测序技术,对长片段的PCR扩增子进行高通量解码分析的软件,利用三代测序技术序列读长较长的优势,LaDecode能够识别目的区域中的各种复杂的突变,并区分检测样本的所有单倍型,该工具特别适用于分析特定区域内多靶点编辑(如启动子区域饱和突变和平铺删除)引起的复杂变异。
SuperDecode具有对用户友好易用的自动化操作界面。此外,HiDecode和LaDecode支持用户自定义barcode序列构建文库或者是单样本的测序文件,具有更加强大的灵活性。除了检测由基因编辑产生的突变,也可以用于检测自然遗传变异,用于基因分型等。
相关论文信息:https://doi.org/10.1016/j.molp.2025.03.002
版权声明:凡本网注明“来源:中国科学报、科学网、科学新闻杂志”的所有作品,网站转载,请在正文上方注明来源和作者,且不得对内容作实质性改动;微信公众号、头条号等新媒体平台,转载请联系授权。邮箱:shouquan@stimes.cn。






