|
|
|
|
|
QB期刊 | 西湖大学王寿文团队综述DNA甲基化与谱系追踪的历史、进展与未来方向 |
|
|
论文标题:DNA methylation meets lineage tracing: History, recent progress, and future directions
期刊:Quantitative Biology
作者:Ruijiang Fu, Mengyang Chen, Shou-Wen Wang
发表时间:28 Oct 2025
DOI:10.1002/qub2.70017
微信链接:点击此处阅读微信文章
近几十年来,谱系追踪技术借助新型基因工程工具得到了快速发展。然而,这些技术因具有侵入性(需向基因组引入外源片段),难以直接应用于人类。虽然内源性DNA突变可用于人体谱系追踪,但其极低的突变率带来了巨大的技术挑战。相比之下,DNA甲基化上的表观突变发生频繁、信息丰富且稳定遗传,加之具有非侵入性,使其成为人类谱系追踪的理想选择。
近日,西湖大学王寿文团队在Quantitative Biology期刊上发表了题为“DNA methylation meets lineage tracing: History, recent progress, and future directions”的展望文章。该文系统综述了DNA甲基化在谱系追踪中的应用历史,重点讨论了MethylTree和EPI-Clone两项技术的发展,并对比了两种方法的优势与局限,同时探讨了未来基于表观突变的谱系追踪工具开发方向。

全文概要
谱系追踪是一种利用独特且可遗传的标记来记录细胞分裂历史的技术,是研究发育、组织稳态、衰老和疾病进程的有力工具。然而,基于基因工程改造的谱系追踪技术大多难以直接应用于人类。虽然体细胞DNA突变已被证明可用于人类谱系追踪,但这些突变在正常细胞中极为罕见。线粒体DNA(mtDNA)突变是另一种选择,但由于其遗传可能受到中性漂变的影响,或经历正向与负向选择,仅有一部分突变能可靠地追踪细胞谱系。此外,mtDNA突变仅在发生大量克隆扩增的系统(如癌症)中表现较好,且其分析受到过多数据噪声的困扰,因此存在一定的局限性。
在哺乳动物中,DNA甲基化主要发生在基因组中的CpG位点,且能在细胞分裂过程中稳定遗传,这使其有望成为高分辨率重建细胞谱系的理想标记。多项研究已表明,DNA甲基化不仅能记录克隆记忆,而且结合群体全基因组测序鉴定的拷贝数变异信息,还可用于推断癌症的演化历史。此外,DNA甲基化在早期胚胎发育中也能记录细胞谱系;在植物种,基于DNA甲基化的表观遗传时钟同样被用于重建系统发育关系。本文将重点介绍基于DNA甲基化进行谱系追踪的MethylTree和EPI-Clone两项技术。
1. MethylTree:首个基于表观突变的通用谱系追踪工具
MethylTree利用两个细胞之间共同检测到的CpG位点来计算它们的Pearson相关系数(图1A)。这解决了海量缺失数据的问题,从而构建出反映这些细胞谱系相似性的相关矩阵。为了处理异质性噪声,MethylTree迭代推断每个细胞的噪声因子,并相应地校正相似性矩阵。研究表明,经过这种噪声清理后,MethylTree的表现显著提升。最后,基于先验细胞类型信息(可来自荧光激活细胞分选(FACS)或联合转录组分析),MethylTree能够去除与不同细胞类型相关的功能性甲基化信号,从而提取出谱系特异性相似性用于下游分析(图1A)。通过这些方法,MethylTree在广泛的生物学条件下以近100%的准确率从单细胞DNA甲基化组重建了细胞谱系,成为首个基于单细胞DNA甲基化的通用谱系追踪工具。
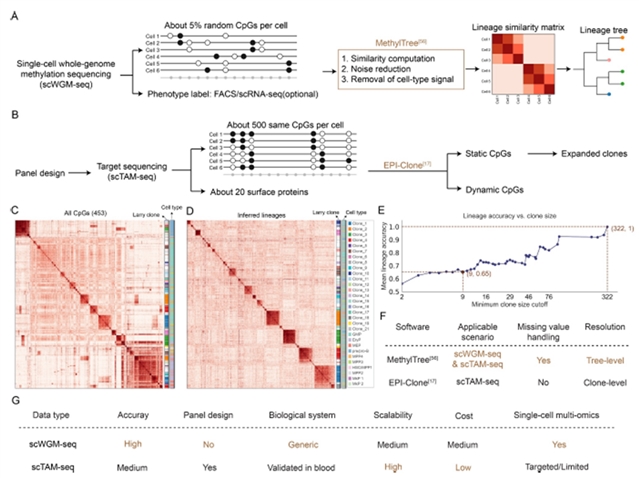
图1. MethylTree 和 EPI‐Clone技术比较
2. EPI-Clone:基于靶向测序的互补方法
EPI-Clone基于靶向单细胞DNA甲基化测序,利用其团队开发的scTAM-seq方法,可在Mission Bio Tapestri平台上对单细胞中数百个CpG位点的甲基化状态进行分析。scTAM-seq能够高效地在不同细胞间对相同CpG位点进行读取,缺失率仅为7%。该方法本质上对数据进行统计检验,将与蛋白标志物差异表达相关的CpG位点鉴定为动态CpG,而将无此类相关性的CpG位点指定为静态CpG。最后,基于静态CpG生成的低维空间,可将细胞聚类为不同克隆(图1B)。需要注意的是,EPI-Clone只能鉴定扩增克隆,且对小克隆的分辨率较低。
3. MethylTree也适用于scTAM-seq数据
MethylTree是一款开箱即用的计算工具包,能够从单细胞DNA甲基化数据中推断细胞谱系,无论数据来自全基因组测序还是靶向扩增。因此,MethylTree也应该适用于scTAM-seq数据。如图1C-E,MethylTree在该靶向数据上的表现与EPI-Clone相当,两者在鉴定大克隆方面表现良好,但均无法检测小克隆。因此,MethylTree为从不同单细胞DNA甲基化数据中推断谱系提供了一种统一的解决方案。
4. MethylTree与EPI-Clone的比较
MethylTree代表全基因组组学方法(图1A),而EPI-Clone代表基于scTAM-seq的靶向方法(图1B)。这两种方法互为补充,但也存在重要差异。首先,MethylTree作为通用计算工具适用于全基因组和靶向数据,而EPI-Clone仅适用于靶向测序。其次,全基因组方法(MethylTree)可直接适用于不同生物学系统,且分辨率和准确性更高;靶向方法(EPI-Clone)在研究新组织时需要仔细选择CpG panel,但该方法更易于扩展且成本更低。此外,靶向scTAM-seq数据专为谱系追踪量身定制,但全基因组测量不仅对谱系追踪有用,还可用于探索性分析表观基因组(即此处指DNA甲基化组)在细胞命运选择中的作用。
5. 基于表观突变的谱系追踪技术优势与未来发展方向
迄今为止,人类非侵入性谱系追踪可通过核DNA突变、线粒体DNA突变或DNA甲基化表观突变来实现。然而,由于线粒体DNA突变的遗传机制复杂,推断的谱系往往不准确;核DNA每次分裂仅积累约1个突变,检测非常困难且成本高昂。相比之下,DNA甲基化每次细胞分裂约积累10,000个表观突变,且可在单细胞分辨率上轻松测量,使其成为理想的谱系记录器。此外,单细胞DNA甲基化组可与其他模态(如转录组)进行联合分析,不仅可以实现单细胞多组学谱系追踪,还能系统性地解析人类的发育与疾病。
未来,开发一种在准确性、可扩展性和成本之间取得良好平衡的基于表观突变的谱系追踪平台将非常有价值。开发更具可扩展性、同时能捕获其他模态的scWGM-seq方法本身也具有重要意义。改进靶向方法同样颇具吸引力,特别是如果它能捕获更多CpG位点以提供更高分辨率的话。此外,从单细胞DNA甲基化中计算提取更多信息(如细胞分裂时间)也将具有重要价值。最后,增加空间模态虽然技术上具有挑战性,但将是实现组织中空间谱系追踪的一个令人兴奋的未来方向。
QB期刊介绍
Quantitative Biology (QB)期刊是由清华大学、北京大学、高等教育出版社联合创办的全英文学术期刊。由高等教育出版社和Wiley双平台出版和发行。QB主要刊登生物信息学、计算生物学、系统生物学、理论生物学和合成生物学的最新研究成果和前沿进展,并为生命科学与计算机、数学、物理等交叉研究领域打造一个学术水平高、可读性强、具有全球影响力的交叉学科期刊品牌。
QB期刊目前已被ESCI, PMC, Scopus, DOAJ, CSCD等国内外重要数据库收录。
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。






