|
|
|
|
|
FIE Na2FeSiO4作为钠离子电池材料:一种计算视角 |
|
|
论文标题:Na2FeSiO4 as a sodium-ion battery material: A computational perspective
期刊:Frontiers in Energy
作者:Ratnasingam Sriraam, Poobalasingam Abiman, Poobalasuntharam Iyngaran, Navaratnarajah Kuganathan
发表时间:14 Oct 2025
DOI: 10.1007/s11708-025-1040-2
微信链接:点击此处阅读微信文章

文章简介
钠离子电池(SIBs)作为锂离子电池(LIBs)的重要补充,因其资源丰富和成本低廉而受到广泛关注。在众多正极材料中,聚阴离子型硅酸盐材料因其结构稳定性和高工作电压展现出巨大潜力。本研究以Na2FeSiO4为研究对象,通过原子尺度模拟系统探讨了其本征缺陷、钠离子迁移路径及掺杂行为,为理解该材料在钠离子电池中的应用提供了理论基础。
研究背景及意义
钠离子电池的发展源于对可持续能源存储需求的日益增长。随着可再生能源如太阳能和风能的普及,高效、安全的储能技术成为支撑智能电网和电动交通的关键。尽管锂离子电池目前占据主导地位,但锂资源的有限性和地理分布不均促使科研人员转向钠基电池系统。钠元素在地壳中储量丰富、分布广泛,使得钠离子电池在大规模储能应用中具有显著经济优势。聚阴离子型正极材料,特别是硅酸盐类化合物,因其开放框架结构、优异的热稳定性和可调节的电化学性能成为研究热点。其中,Na2FeSiO4凭借其高理论容量(276 mAh/g)和环保特性备受关注。然而,该材料存在多种晶相,其电化学性能与晶体结构密切相关,尤其是单斜相的研究尚不充分。因此,通过计算模拟深入解析Na2FeSiO4的缺陷化学和离子传输机制,对优化其电池性能具有重要意义。
主要研究内容
本研究采用经典势函数模拟和密度泛函理论(DFT)相结合的方法,系统分析了Na2FeSiO4单斜相的结构特性、缺陷形成能、钠离子迁移路径及掺杂效应。首先,利用通用晶格程序(GULP)优化晶体结构,结果显示计算得到的晶格参数与实验值高度吻合,误差小于1%,验证了模拟方法的可靠性。电荷密度分布表明电子主要局域于氧原子周围,而态密度(DOS)分析显示DFT+U方法能更准确地描述Fe 3d轨道的电子局域性,带隙约为3 eV,与实验值一致。Bader电荷分析进一步揭示了材料中离子的混合离子-共价键合特性。
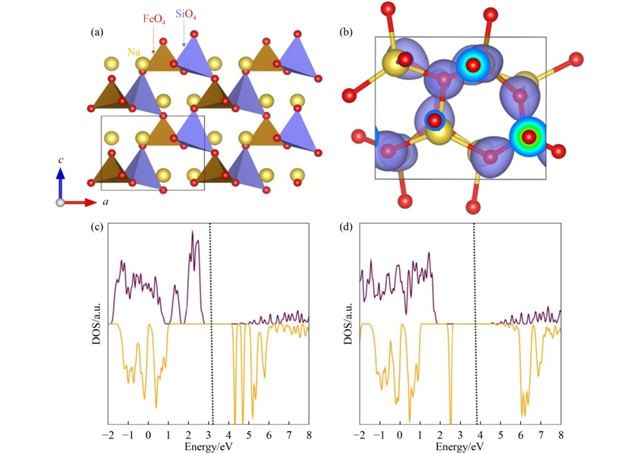
图1 Na2FeSiO4的结构和电子特性
在缺陷研究方面,本征点缺陷的形成能被系统计算。结果表明,钠弗伦克尔缺陷(Na Frenkel)的形成能最低(1.71 eV),表明材料中钠空位易于形成,有利于钠离子通过空位机制迁移。此外,反位缺陷(如Na/Fe反位)在团簇形式下能量更低(1.09 eV),说明缺陷聚集在热力学上更有利。钠离子迁移路径分析识别出四条可能的跃迁路径,其中两条关键路径的激活能垒分别为0.38 eV和0.41 eV,构成了长程扩散通道。这一数值低于许多同类硅酸盐材料,如Na2MnSiO4(0.81 eV)和Li2FeSiO4(0.83 eV),表明Na2FeSiO4具有较好的离子电导率。

图2 缺陷反应能
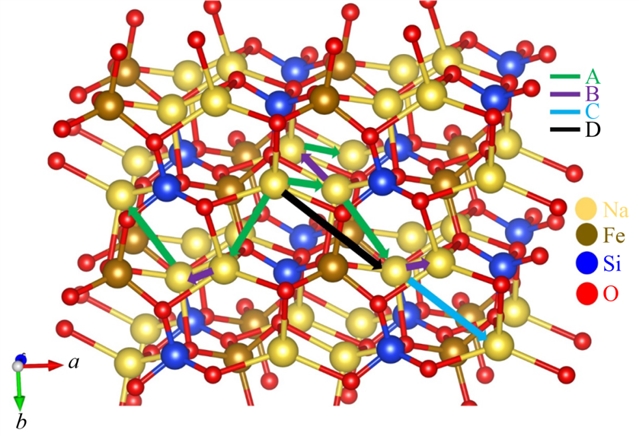
图3 硅酸亚铁钠Na2FeSiO4中的钠离子迁移路径
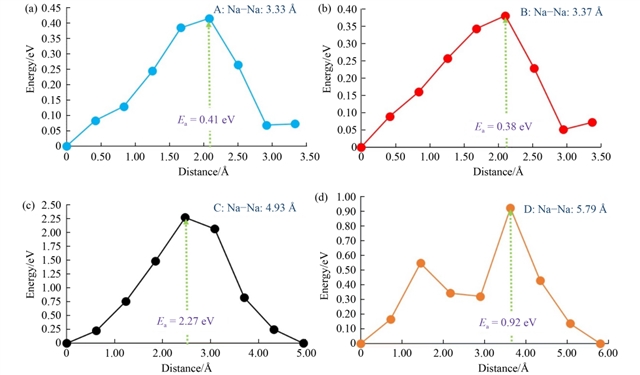
图4 局域钠离子跳跃的能量分布曲线及活化能
掺杂研究聚焦于不同价态元素在Na、Fe和Si位的取代行为。对于单价掺杂,K在Na位的溶解能最低(0.13 eV),其引入未引起显著结构畸变或电子结构变化,总态密度显示费米能级仍位于带隙中。二价掺杂中,Zn在Fe位的溶解能最低(0.47 eV),其原子尺寸与Fe2+接近,掺杂后晶体结构保持完整,且未引入带隙态。三价掺杂在Fe位时,Ga表现出最低溶解能(3.78 eV),并能促进钠空位形成,有助于增强离子电导;而在Si位掺杂时,Al的溶解能相对较低(5.71 eV),并通过引入间隙钠离子提高材料总钠含量。四价掺杂中,Ge在Si位的溶解能最低(2.05 eV),且其掺杂使费米能级向价带移动,可能提升电子电导。所有优选掺杂剂均通过DFT分析了其电子结构,证实它们未破坏材料的绝缘本性,但Ge等元素可能对电化学性能产生积极调制。
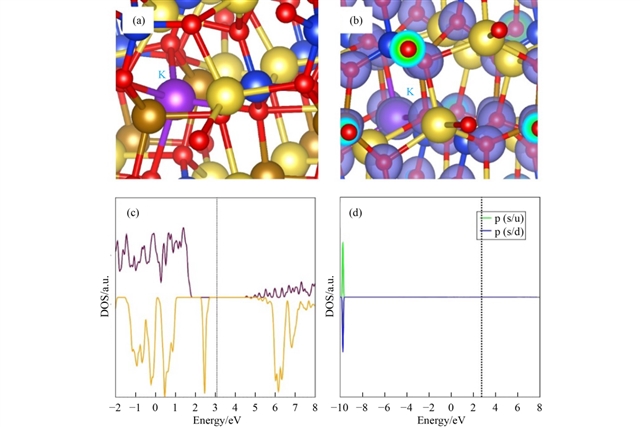
图5 K掺杂对Na2FeSiO4的影响
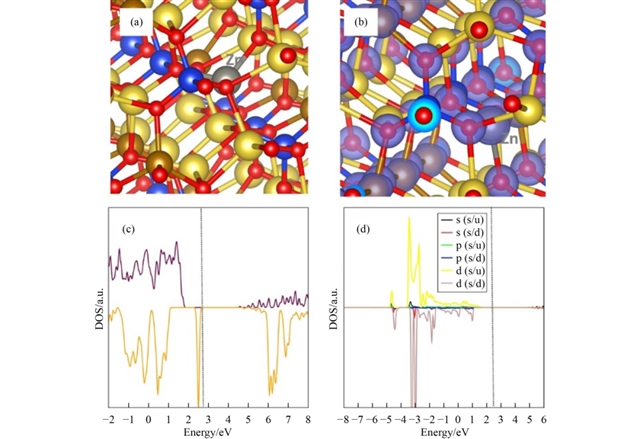
图6 Zn掺杂对Na2FeSiO4的影响,
研究结论
综上所述,本研究通过原子尺度模拟揭示了Na2FeSiO4作为钠离子电池正极材料的潜力。其低钠弗伦克尔缺陷形成能和适中的钠离子迁移能垒表明该材料具备良好的离子传输性能。掺杂研究筛选出K、Zn和Ge分别为Na、Fe和Si位的最优等价位掺杂剂,而Ga和Al则分别通过促进空位形成和增加钠含量优化电极行为。这些发现为实验制备和性能优化提供了理论指导。未来研究应侧重于实验验证计算预测、探索共掺杂策略以及温度对缺陷动力学的影响,以推动Na2FeSiO4在实际电池系统中的广泛应用。
原文信息
Na2FeSiO4 as a sodium-ion battery material: A computational perspective
Ratnasingam Sriraam¹, Poobalasingam Abiman1, Poobalasuntharam Iyngaran1, Navaratnarajah Kuganathan2
Author information:
1. Department of Chemistry, University of Jaffna, Thirunelvely, Jaffna 40000, Sri Lanka
2. Department of Materials, Imperial College London, London SW7 2AZ, UK
Abstract:
Polyanionic silicate-based cathode materials have attracted considerable attention due to their intrinsic structural stability, strong thermal and chemical resistance, and ability to achieve high operating voltages through the inductive effects of polyanion groups. In this study, atomistic simulations were conducted to explore the energetics of intrinsic point defect formation, Na-ion migration pathways, and dopant incorporation in Na2FeSiO4, providing key insights into its viability as a cathode material for sodium-ion batteries (SIBs). Among the native defects, the Na Frenkel pair exhibited the lowest formation energy, suggesting a natural preference for vacancy-mediated Na-ion migration. The calculated migration energy barriers of 0.38 and 0.41 eV further support the material’s capability for efficient sodium-ion transport. Doping analysis identified K, Zn, and Ge as the most favorable isovalent dopants at the Na, Fe, and Si sites, respectively, while Ga showed a strong tendency to substitute at Fe sites and facilitate Na-vacancy formation. Furthermore, Al substitution at the Si site was found to increase the overall sodium content in the lattice. The electronic structure of these promising dopants was further investigated using density functional theory (DFT), offering deeper insights into their influence on the electrochemical behavior of Na2FeSiO4.
Keywords:
Batteries; cathode; defects; density functional theory (DFT) ; diffusion
Cite this article:
Ratnasingam Sriraam, Poobalasingam Abiman, Poobalasuntharam Iyngaran, Navaratnarajah Kuganathan. Na2FeSiO4 as a sodium-ion battery material: A computational perspective. Front. Energy
DOI:10.1007/s11708-025-1040-2
扫描二维码,阅读原文

期刊简介
Frontiers in Energy是中国工程院院刊能源分刊,高教社Frontiers系列期刊之一。由中国工程院、上海交通大学和高等教育出版社共同主办。翁史烈院士和倪维斗院士为名誉主编,中国工程院院士黄震、周守为、苏义脑、彭苏萍担任主编。加拿大皇家科学院、加拿大工程院、中国工程院外籍院士张久俊,美国康涅狄格大学校长、教授Radenka Maric,上海交通大学教授Nicolas Alonso-Vante和巨永林担任副主编。

Frontiers in Energy已被SCIE、EI、Scopus、CAS、INSPEC、Google Scholar、CSCD、中国科技核心期刊等数据库收录。根据《期刊引证报告》,本刊2024年影响因子为6.2,在“ENERGY & FUELS”学科分类中位列55位(55/182),处于JCR Q2区。2024年度CiteScore为6.9,在“Energy”领域排名#77/299;2025年即时影响因子为7.1,即时CiteScore为9.0(数据截至2025年12月20日)。
Frontiers in Energy出版能源领域原创研究论文、综述、展望、观点、评论、新闻热点等。选文注重“前沿性、创新性和交叉性”,涉及领域包括:能源转化与利用,可再生能源,储能技术,氢能与燃料电池,二氧化碳捕集、利用与封存,动力电池与电动汽车,先进核能技术,智能电网和微电网,新型能源系统,能源与环境,能源经济和政策。
Frontiers in Energy免收版面费,并为录用的文章提供免费语言润色服务,以确保出版质量。第一轮平均审稿周期30天,从审稿到录用平均60天。
更多信息请访问:
http://journal.hep.com.cn/fie(国内免费开放)
https://link.springer.com/journal/11708
联系我们:
FIE@sjtu.edu.cn, (86) 21-62932006
qiaoxy@hep.com.cn, (86) 10-58556482
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。






