
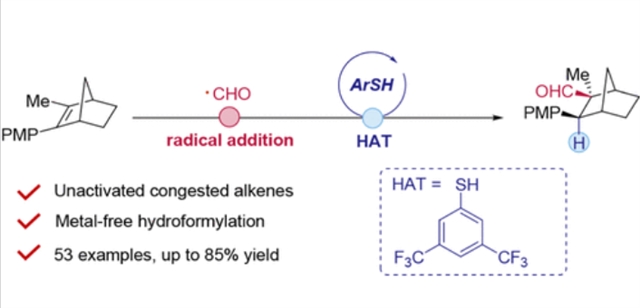
近日,中国科学院上海有机化学研究所陈以昀团队研究了噻吩催化非活化位阻烯烃的自由基氢甲酰化。相关论文于2025年8月21日发表在《美国化学会志》上。
由于过渡金属催化中不可抑制的位阻应变,传统的氢甲酰化仍然无法得到位阻密集的烯烃──药物和内质中间体中的泛位烯。
研究组报道了一种噻吩催化的自由基氢甲酰化反应,通过一个协同系统:稳定的α-氯N-甲氧基酞酰亚胺(甲酰基前体)和一种定制的噻吩HAT催化剂,克服了双重空间和电子限制。这种无金属策略实现了前所未有的非活化、三取代和四取代烯烃(包括富电子苯乙烯和脂肪烯烃系统)的氢甲酰化,具有高化学和区域选择性。
主要进展包括:1)四取代苯乙烯的首次氢甲酰化,生成具有季中心的β-芳基醛(产率高达74%);2)噻吩降低了8.3 kcal/mol的HAT势垒,使位阻堵塞下的催化转化成为可能。机制研究证实了甲酰基自由基途径,而合成气(CO/H2)消除为合成医学相关基序(如肟醚)和百合衍生物等内源性靶标建立了一个安全、可持续的平台。
附:英文原文
Title: Thiophenol-Catalyzed Radical Hydroformylation of Unactivated Sterically Hindered Alkenes
Author: Yisicheng Wang, Panpan Bao, Xiaojuan Dong, Yu Lan, Yiyun Chen
Issue&Volume: August 21, 2025
Abstract: Sterically congested alkenes─ubiquitous in pharmaceuticals and industrial intermediates─remain inaccessible to classical hydroformylation due to prohibitive steric strain in transition-metal catalysis. Here, we report a thiophenol-catalyzed radical hydroformylation that overcomes dual steric and electronic constraints through a synergistic system: bench-stable α-chloro N-methoxyphthalimides (formyl precursors) and a tailored thiophenol HAT catalyst. This metal-free strategy achieves unprecedented hydroformylation of unactivated, tri-, and tetrasubstituted alkenes─including electron-rich styrenes and aliphatic alkene systems─with high chemo- and regioselectivity. Key advances include: 1) the first hydroformylation of tetrasubstituted styrenes, delivering β-aryl aldehydes with quaternary centers (up to 74% yield); 2) thiophenol lowers the HAT barrier by 8.3 kcal/mol to enable catalytic turnover under steric congestion. Mechanistic studies confirm a formyl radical pathway, while syngas (CO/H2) elimination establishes a safe, sustainable platform for synthesizing medicinally relevant motifs (e.g., oxime ethers) and industrial targets like Lilial derivatives.
DOI: 10.1021/jacs.5c07415
Source: https://pubs.acs.org/doi/abs/10.1021/jacs.5c07415
JACS:《美国化学会志》,创刊于1879年。隶属于美国化学会,最新IF:16.383
官方网址:https://pubs.acs.org/journal/jacsat
投稿链接:https://acsparagonplus.acs.org/psweb/loginForm?code=1000