|
|
|
|
|
研究人员揭示SOD1病理突变体激活细胞铁死亡并诱发家族遗传型ALS新机制 |
|
|
北京时间2024年11月1日,《科学进展》(Science Advances)以长文(Research Article)形式在线发表了武汉大学团队和中国科学院上海有机所交叉中心团队的最新研究成果。
研究人员首次在原子水平上解析了铜锌超氧化物歧化酶(SOD1)病理突变体H46R和G85R淀粉样纤维的高分辨率冷冻电镜结构(3.11 Å和2.97 Å),揭示了H46R和G85R淀粉样纤维对神经细胞铁死亡激活调控,并诱发家族遗传型萎缩侧索硬化症(ALS)新机制,为发展新的针对SOD1突变体纤维的ALS治疗药物奠定了基础。
武汉大学生命科学学院梁毅教授和中国科学院上海有机化学研究所交叉中心刘聪研究员为该论文的共同通讯作者;武汉大学生命科学学院博士后王利强、中国科学院上海有机化学研究所交叉中心博士生马烨阳和武汉大学生命科学学院博士生张沐雅为共同第一作者;武汉大学土木建筑工程学院王正直教授、深圳市人民医院神经内科邹良玉教授等参与了该论文的研究工作,武汉大学科研公共服务条件平台冷冻电镜机组特聘高级工程师李丹阳和实验员李香凝在数据收集工作中提供了重要协助。
ALS又称渐冻症或渐冻人症,是一种进行性的、致命的神经退行性疾病。SOD1病理突变体在中枢神经系统的运动神经元形成纤维样聚集体是ALS的重要病理特征之一。SOD1编码基因sod1是最早被发现的同时又是第二常见的与ALS有关的基因,迄今为止已经发现引起家族遗传性ALS的SOD1突变体多达208种,然而,这些SOD1突变体诱发家族遗传型ALS的分子机制仍不清楚。
在对ALS患者的中央神经系统的解剖中科学家曾多次鉴定到SOD1的错误构象和聚集物,从SOD1突变的ALS转基因小鼠脊髓中也可分离得到SOD1的纤维样聚集体,然而尽管大量证据显示了SOD1异常聚集与神经退行性疾病发生间存在密切关联,目前的研究仍不能确切阐释SOD1聚集在ALS的病理损伤中究竟发挥着怎样的效应,又是通过什么机制形成并影响着发病进程。很多SOD1病理突变体仍然保持着近似野生型SOD1的酶活性,这意味着酶活性的丧失并非是SOD1产生神经毒性的主要原因,SOD1很可能通过形成异常构象或聚集获得了毒性,而突变则加剧了蛋白质发生错误折叠和异常聚集的倾向。
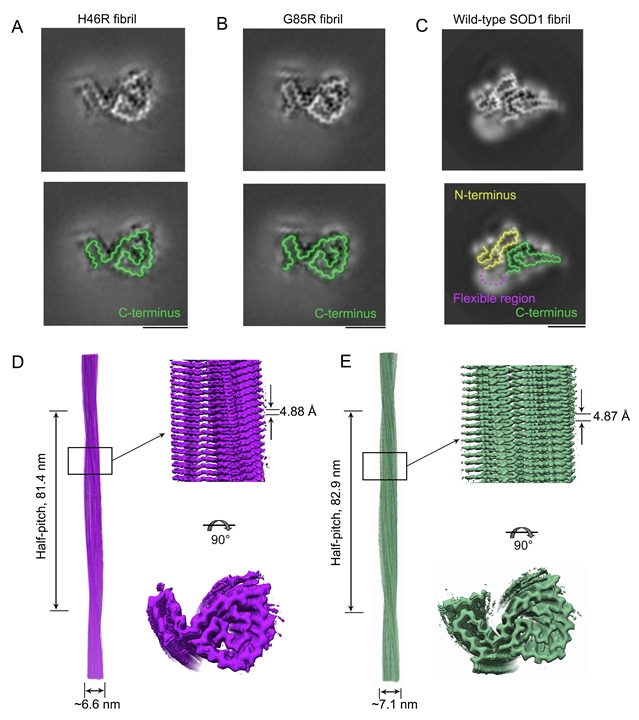
图1:SOD1病理突变体H46R和G85R的纤维结构具有相似的仅由C端片段组成的纤维核心,与在N端和C端间形成了稳定盐桥作用的野生型纤维结构大不相同
在野生型朊蛋白及其病理突变体E196K淀粉样纤维和野生型铜锌超氧化物歧化酶(SOD1)淀粉样纤维冷冻电镜结构及功能前期工作(2020年6月8日、2021年9月9日、2022年6月17日和2023年12月8日分别发表于《自然¾结构与分子生物学》(Nat. Struct. Mol. Biol. 2020, 27, 598-602)、《科学进展》(Sci. Adv. 2021, 7, eabg9676)、《自然—通讯》(Nat. Commun. 2022, 13, 3491)和《自然—通讯》(Nat. Commun. 2023, 14, 8131))中,梁毅与刘聪团队合作解析了全长野生型朊病毒蛋白纤维、全长E196K纤维和类朊病毒蛋白野生型SOD1纤维的结构,首次在原子水平上揭示了朊蛋白由细胞型朊蛋白(PrPC)向病理型朊病毒蛋白(PrPSc)结构转变的机制,揭示了朊病毒蛋白病理聚集多态性的分子机制,揭示了ALS致病蛋白质SOD1构象转化分子机制,揭示了miRNA对PrPC相分离、肌肉细胞自噬及分化调控机制。H46R和G85R是两种显著影响金属离子结合的病理突变体,然而SOD1病理突变体到底是怎么激活调控神经细胞铁死亡并诱发家族遗传型ALS的仍是困扰科学界的谜题。
“冷冻电镜(Cryo-EM)技术为蛋白质错误折叠与神经退行性疾病带来革命性的进展;解析SOD1病理突变体纤维的高分辨率结构将为阐释ALS发病的分子机制起到极大的推动。”论文指出。
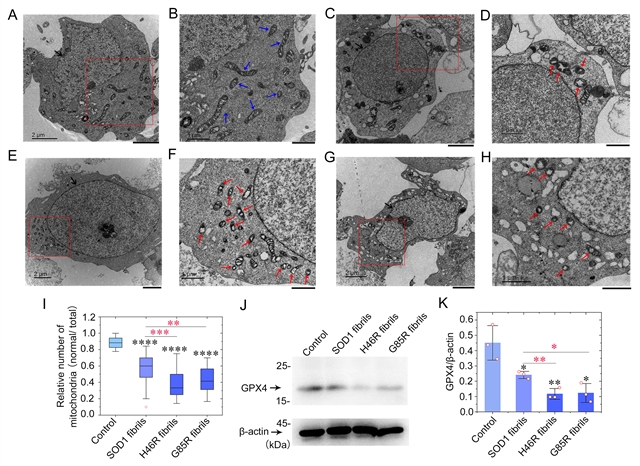
图2:SOD1病理突变体H46R和G85R的纤维种子显著激活受GPX4调节的神经细胞铁死亡
梁毅介绍,为了回答“SOD1突变到底是怎么诱发家族遗传型ALS的”这一科学问题,在这项研究中,研究人员通过激光共聚焦显微和免疫印迹等方法发现,与野生型SOD1纤维相比,这些突变体纤维具有更强的诱导细胞内源SOD1聚集和线粒体功能紊乱的能力,显著激活受谷胱甘肽过氧化物酶4(GPX4)调节的神经细胞铁死亡。GPX家族是在细胞氧化还原通路中位于SOD家族下游的过氧化物酶,GPX4可以清除膜脂过氧化氢产物,更是一种铁死亡的重要抑制因子,近年来被用作判断细胞发生铁死亡的关键指标。该研究的实验结果提示,这些突变体纤维种子可能通过诱导聚集效应触发一系列复杂的氧化损伤通路,乃至激发了铁死亡途径,最终诱发家族遗传型ALS。
通过进一步研究,研究团队发现,这两种显著影响金属离子结合的病理突变体H46R和G85R的纤维结构具有相似的仅由C端片段组成的纤维核心,与在N端和C端间形成了稳定盐桥作用的野生型纤维结构大不相同,异硫氰酸胍解聚实验发现这两种突变体纤维的稳定性相比野生型显著降低。在G85R聚集过程中,单股原纤维通过病理突变位点的Arg85和Asp101之间形成的盐桥发生相互作用,形成一个亲水空腔,而在H46R聚集过程中,单股原纤维则通过Asn86和Asp101之间形成的氢键发生相互作用,形成一个亲水空腔。此前研究表明,SOD1氨基酸位点突变会带来的自由能形貌变化和活化构象扰动,那么该工作解析到的不同突变体生长纤维的结构多样性或许是一系列物理化学性质变化的下游事件。
在细胞水平实验中,H46R、G85R的纤维种子表现出比野生型种子更强的细胞毒性并造成了更显著的线粒体损伤,用野生型SOD1、H46R或G85R纤维的种子处理稳转FLAG-野生型SOD1的HEK-293T细胞均可以诱导内源过表达SOD1的共聚集,并从细胞裂解物中检测到FLAG阳性Sarkosyl不溶性沉淀,但两种突变体纤维的诱导聚集能力明显强于野生型。
这项工作的研究结果表明,H46R和G85R纤维核心分别由其C端的85-153和82-153组成,H46R和G85R纤维都由单股原纤维以左手螺旋的方式缠绕而成,纤维核心的部分疏水氨基酸侧链向内折叠形成1个长的疏水空腔,起到了稳定淀粉样纤维结构的作用。这两种病理突变使得SOD1丧失了大部分结合铜离子和锌离子的能力,导致SOD1纤维的结构发生重排,H46R和G85R纤维核心的部分亲水氨基酸侧链向内折叠形成3个亲水空腔,使得这些突变体纤维相较野生型纤维具有更不稳定的构象。
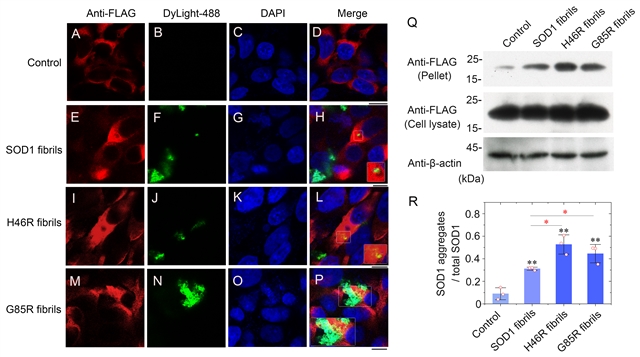
图3:SOD1病理突变体H46R和G85R的纤维种子具有更强的诱导细胞内源SOD1聚集(“模板折叠”)的能力
该研究首次揭示了H46R纤维、G85R纤维和野生型SOD1纤维之间的结构差异,在原子水平上揭示了SOD1病理突变体H46R和G85R淀粉样纤维激活细胞铁死亡的机制,证明金属离子结合区域的两种病理突变都能够破坏SOD1纤维中重要的相互作用(例如盐桥),使得这些病理突变体纤维结构形成了彼此类似但又完全不同于野生型SOD1纤维的构象,突显了具有相似功能的SOD1病理突变体可能表现出相似的纤维结构的重要性,并使得发展新的针对SOD1突变体纤维及其激活的铁死亡的ALS治疗药物成为可能。(来源:科学网)
相关论文信息:DOI: https://www.science.org/doi/10.1126/sciadv.ado8499






