|
|
|
|
|
南方科技大学张绪穆团队实现前列腺素类化合物高效不对称合成 |
|
|
前列腺素(Prostaglandin, PG)是一类广泛存在于动物体内的自体活性物质,介导体内多种生理功能。前列腺素类化合物具有非常高的成药性,目前,在全球范围内有超过20种前列腺素类上市药物。由于前列腺素类化合物独特的结构和广泛的生物活性,发展前列腺素的高效合成方法具有非常重要的意义。在过去的50多年间,前列腺素一直是合成化学家的重要目标分子。
目前,南方科技大学化学系张绪穆、陈根强研究团队以自主开发的烯炔环异构化反应和不对称氢化反应为关键步骤实现了前列腺素类化合物的高效不对称合成,极大地提高了合成效率。
相关研究成果以“Concise, scalable and enantioselective total synthesis of prostaglandins”为题,于北京时间2021年5月27日晚23时发表在Nature Chemistry上。
1934年,前列腺素被von Euler等人首次发现,当时认为此物质可能是由前列腺所分泌,因此命名为前列腺素。1957年Bergström及其瑞典同事Samuelsson分离出了PGF1和PGF2α的纯品,并在后续研究中阐明了前列腺素的结构和代谢过程,有关前列腺素药物的研究在随后的20世纪60年代达到高潮。1982年,Bergström与Samuelsson和John Vane三位科学家因在前列腺素及有关生物活性物质的研究方面的卓越贡献共同获得了诺贝尔生理学和医学奖。
PGF2α是前列腺素家族中最复杂的化合物,其核心结构为具有四个连续手性中心的环戊烷骨架和两个侧链(图1a)。1964年,E. J. Corey首次实现了PGF2α的全合成,随后,众多著名的有机化学家如Woodward、Stork、Noyori、Danishefsky、Aggarwal、Baran、Grubbs、陈芬儿等也在前列腺素的合成方面做出了许多重要的研究工作。尽管前列腺素的合成取得了重要进展,已经开发了多条合成路线,但是,绝大多数的合成路线不适用于工业化合成,目前前列腺素类药物的工业合成仍然主要依赖Corey内酯(10),Corey内酯可以从环戊二烯9出发经过9步合成得到,从Corey内酯出发仍需要多步反应才能够得到前列腺素,有些前列腺素分子甚至需要超过10步来合成(图1b)。在该研究工作中,作者设计合成了一个合成前列腺素的关键中间体15,从该中间体出发仅需要两步就可以合成得到前列腺素化合物,该中间体可以通过简单易得的β-酮酰胺11经过4步合成得到(图1c)。
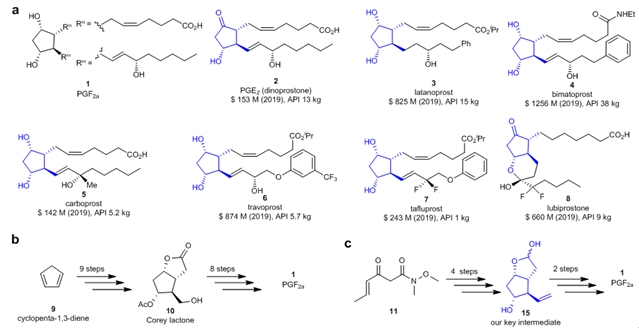
图1:a. 一些代表性的前列腺素类药物的结构;b. Corey小组前列腺酸的合成方法(Corey内酯为关键中间体);c.张绪穆团队开发的前列腺素的合成方法(化合物15为关键中间体)
前列腺素不对称合成中的主要难点在于其核心环戊烷骨架中四个连续手性中心的精准构建以及两个侧链的高效引入。2000年,张绪穆研究团队报道了首例铑催化的1,6-烯炔的环异构化反应,可以实现手性五元环化合物的高效构建,对于大多数底物,采用常见的BINAP作为手性配体,该反应就可以获得非常高的对映选择性。2014,该反应被人名反应收录(张-烯炔环异构化反应),成为为数不多的几个以华人姓氏命名的人名反应(Li, J. J. Zhang enyne cycloisomerization in Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications 652–653, Springer, 2014)(图2)。2016年,张绪穆研究团队设计合成了一类基于二茂铁骨架的手性三齿配体f-amphox (L1),该配体与铱的络合物在简单酮的不对称氢化中表现出优异的活性和对映选择性(up to 1,000,000 TON, up to >99% ee),甚至在无溶剂条件下反应也能够高效进行(Org. Lett. 2016, 18, 2938-2941)。
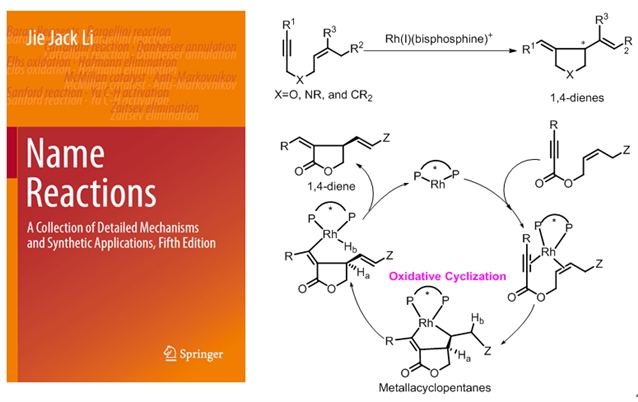
图2:张-烯炔环异构化反应及其机理
研究团队设想以铑催化的不对称烯炔环异构化反应为关键步骤构建手性环戊烷骨架,进而实现前列腺素PGF2α的不对称合成。他们对前列腺素PGF2α进行了逆合成分析(图3),发现目标化合物PGF2α (1)可以通过关键中间体15,经过Grubbs烯烃交叉复分解反应和Wittig反应合成得到,而关键中间体15则可以通过1,6-烯炔底物13的不对称环异构化反应和后续的还原得到,1,6-烯炔底物13可以通过化合物12和化合物18的锂盐的加成反应合成得到,化合物12则可以通过简单易得的β-酮酰胺11的不对称氢化和硅基保护得到。
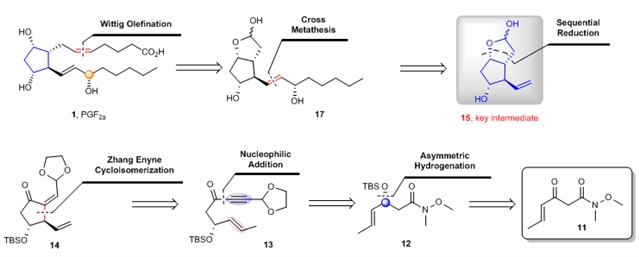
图3:前列腺素的逆合成分析
基于上述逆合成分析,作者以β-酮酰胺11为起始原料,对前列腺素PGF2α的合成进行了探索(图4)。采用张绪穆研究团队前期开发的f-amphox (L1)为手性配体,β-酮酰胺11在铱催化下可以被对映选择性还原并以70%的收率和94%的对映选择性得到化合物12,Weinreb酰胺类化合物12可以和化合物18的锂盐发生加成反应顺利转化为1,6-烯炔化合物13。以铑为催化剂以及(S)-BINAP为手性配体,1,6-烯炔化合物13发生不对称烯炔环异构化反应,能够以85%的收率、98%的对映选择性和>20/1的非对映选择性得到化合物14。化合物14中的共轭双键和羰基可以分别被二苯基硅氢和三乙基硼氢化锂还原,通过调节还原产物的后处理方式可以选择性地得到关键中间体15或16。中间体16的稳定性大于中间体15,更适合大量合成。中间体16在盐酸的处理下也可以转化为中间体15。
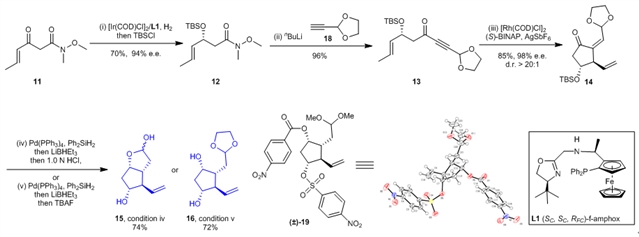
图4:前列腺素关键中间体15和16的高效合成
作者也分别针对不同前列腺素具有的不同侧链,设计了多种高效的合成路线。其中,侧链20,29的合成同样使用了张绪穆研究团队前期自主开发的不对称氢化反应。含有手性叔醇结构的侧链32主要用到Sharpless不对称环氧化反应。而含有芳环结构的侧链37,38则是由廉价易得的环氧氯丙烷为起始原料制备得到。
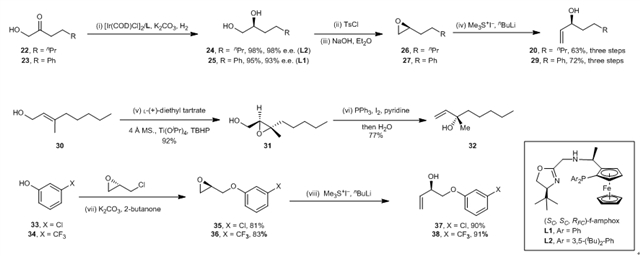
图5:前列腺素侧链的合成
在顺利合成得到关键中间体15和多种侧链后,作者对前列腺素的最终合成进行了尝试(图6)。关键中间体15在Hoveyda-Grubbs二代催化剂的作用下可以和手性醇中间体20发生Grubbs交叉复分解反应顺利得到化合物17,化合物17再经过一步经典的Wittig反应就可以15%的总收率顺利得到前列腺素PGF2α(1)。采用类似的合成路线,拉坦前列素(latanoprost,3)、卡波前列素(carboprost,5)和氯前列烯醇(cloprostenol,20)也能够分别以5.7%、23%以及19%的总收率分别合成得到。值得一提的是,氟前列醇(fluprostenol, 21)可以以27%的总收率和23克的规模合成得到,氟前列醇(fluprostenol, 21)经过一步酯化反应就可以得到曲伏前列素(travoprost, 6)。此外,地诺前列酮(dinoprostone或PGE2, 2)也可以从化合物11出发经过7步反应形式合成得到。
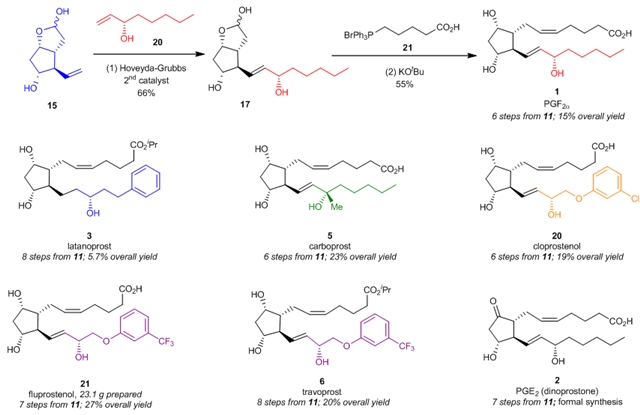
图6:前列腺素的最终合成
最后,张绪穆、陈根强研究团队设计、合成了一种合成前列腺素的关键中间体15和16,并以自主开发的烯炔环异构化反应和不对称氢化为关键步骤实现了该中间体的高效合成。基于该中间体仅需要两步就可以合成得到前列腺素化合物。该合成方法具有原子经济性、步骤经济性和氧化还原经济性等特点,为前列腺素相关药物的合成提供了极大的便利,不仅有望有效降低前列腺素相关药物的生产成本,而且对前列腺素相关药物的研发具有非常重要的意义。
南方科技大学化学系张绪穆和前沿与交叉科学研究院陈根强为本文共同通讯作者,南方科技大学张绪穆教授和新加坡国立大学卢一新教授的联合培养博士研究生张芙豪为本文第一作者。该项研究工作得到了国家自然科学基金、广东省重点领域研发计划项目、广东省普通高校创新团队、深圳市科技创新委员会、深圳市诺奖实验室等项目的资金支持。(来源:科学网)
相关论文信息:https://doi.org/10.1038/s41557-021-00706-1






