近日,中山大学肿瘤防治中心教授潘志忠/林俊忠团队首次揭示了PKM2 K206位点乳酸化修饰驱动结直肠癌血管生成拟态并介导贝伐珠单抗耐药的分子机制,为提高结直肠癌抗血管生成治疗疗效提供了新的治疗靶点和联合治疗策略。相关成果发表于《癌症研究》(Cancer Research)。
 相关研究机制示意图。研究团队供图
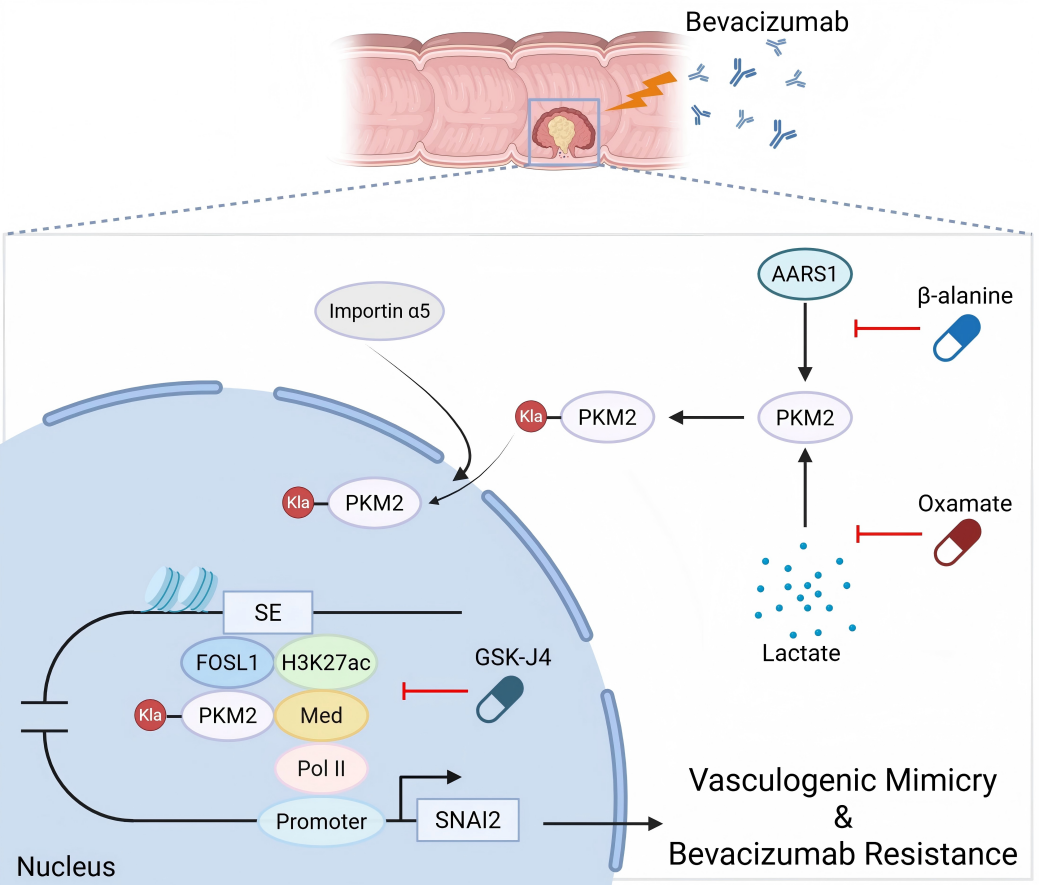
相关研究机制示意图。研究团队供图
结直肠癌是全球最常见的消化道恶性肿瘤之一。以贝伐珠单抗为主的抗血管生成靶向治疗联合化疗,是晚期结直肠癌重要的标准治疗手段。然而,高达60%—70%的患者在治疗过程中出现原发或继发性耐药,导致治疗失败,成为制约疗效的瓶颈。血管生成拟态是肿瘤细胞自主形成的替代性供血通道,是抗血管生成治疗耐药的重要因素,但其分子调控机制尚未完全阐明。
针对这一临床难题,研究团队首先建立了贝伐珠单抗耐药的结直肠癌患者源异种移植(PDX)模型,结合蛋白质组学和乳酸化修饰组学分析发现,耐药肿瘤组织中整体乳酸化修饰水平显著升高,其中糖酵解关键酶PKM2的K206位点乳酸化修饰尤为突出。
AARS1是催化该位点乳酸化的关键乳酰基转移酶,在缺氧微环境下,二者相互作用增强,导致PKM2 K206乳酸化水平显著上调。进一步研究发现,结直肠癌细胞中PKM2 K206乳酸化水平上调可促进肿瘤细胞干性增强及血管生成拟态形成,而抑制其乳酸化则可抑制血管生成拟态形成,进而提高贝伐珠单抗治疗的敏感性。
机制研究显示,PKM2 K206乳酸化并非影响其糖酵解酶活性,而是通过促进importin α5的招募,介导PKM2入核(该过程独立于经典磷酸化通路),发挥非经典功能。入核后的PKM2与转录因子FOSL1结合,共同促进超级增强子形成,激活SNAI2、MYC、MAZ等与肿瘤干细胞特性相关的靶基因转录,最终驱动血管生成拟态形成。换言之,乳酸化修饰为肿瘤细胞开辟了一条“备用供血线路”,使其绕开贝伐珠单抗的抗血管作用而持续增殖,导致治疗抵抗。
在患者源类器官和PDX等临床前模型中,使用草氨酸盐(LDH抑制剂)、β-丙氨酸(AARS1抑制剂)靶向抑制PKM2 K206乳酸化,或使用GSK-J4破坏FOSL1依赖的超级增强子形成,均能显著抑制血管生成拟态,并与贝伐珠单抗产生协同抗肿瘤效果,显著提升治疗响应率。
该研究首次明确了非组蛋白乳酸化修饰与结直肠癌抗血管生成治疗耐药的相关性,揭示了“乳酸-PKM2 K206乳酸化-PKM2入核-FOSL1超级增强子-血管生成拟态”这一全新信号轴,阐明了代谢重编程通过翻译后修饰调控肿瘤转录重编程的分子机制。
该发现不仅为理解结直肠癌耐药提供了新视角,也提出了靶向PKM2乳酸化的联合治疗策略,为晚期耐药结直肠癌患者带来了新的治疗希望。
相关论文信息:https://doi.org/10.1158/0008-5472.CAN-25-3520
版权声明:凡本网注明“来源:中国科学报、科学网、科学新闻杂志”的所有作品,网站转载,请在正文上方注明来源和作者,且不得对内容作实质性改动;微信公众号、头条号等新媒体平台,转载请联系授权。邮箱:shouquan@stimes.cn。






