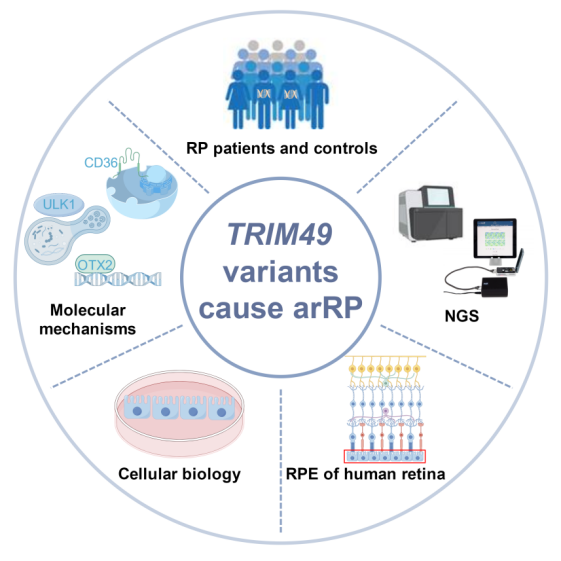
中山大学中山眼科中心教授张清炯、研究员申煌煊团队首次发现TRIM49基因的双等位基因变异是导致常染色体隐性遗传视网膜色素变性(RP)的新病因,并揭示其通过调控ULK1介导的自噬和光感受器外节吞噬,维持视网膜色素上皮(RPE)细胞稳态的全新机制。相关成果近日发表于《先进科学》。
RP是一组以进行性光感受器细胞及RPE细胞变性死亡为特征的常见难治性致盲眼病,其致病基因及发病机制尚未完全明确。RPE细胞堪称维持光感受器细胞功能与存活的关键“后勤保障部队”,其核心功能之一是精准吞噬并消化脱落的光感受器外节碎片。一旦该功能受损,碎片将在视网膜下堆积,最终引发光感受器死亡和视力丧失。
自噬作为细胞内部的“清洁回收系统”,对维持RPE细胞健康及其吞噬功能起着至关重要的作用。然而,目前对于RPE细胞中自噬的具体调控机制,我们仍知之甚少。鉴于RP作为单基因病模型所具备的特性,它成为研究RPE自噬机制的重要突破口。
 相关研究示意图。研究团队供图
相关研究示意图。研究团队供图
?
研究团队通过对两名无血缘关系的RP先证者以及7283例内部对照人群的全外显子组测序数据进行比对分析,在排除已知基因致病变异后,成功锁定了一个新的候选基因——TRIM49。值得一提的是,其中一个家系存在单亲二倍体这一遗传特殊现象。TRIM49是一个参与自噬的灵长类特异性基因,在啮齿类等常见实验动物中并无同源基因。研究发现,TRIM49在人视网膜的转录水平最高,且其蛋白水平特异性表达于人视网膜的RPE细胞。
为进一步明确致病原因,研究团队构建了TRIM49基因缺陷的RPE细胞模型。通过该模型发现,TRIM49基因缺陷会导致RPE细胞出现一系列异常,包括活性氧水平升高、线粒体膜电位下降、ATP合成减少、细胞凋亡增加以及增殖减弱等,这表明TRIM49是维持RPE细胞健康不可或缺的因子。
研究显示,TRIM49缺陷的RPE细胞吞噬光感受器外节的能力显著下降。其深层机制在于,TRIM49缺失会导致吞噬关键受体及其下游信号分子的表达下调,进而阻碍吞噬过程中的“识别”与“内吞”环节。研究发现,TRIM49是RPE细胞自噬流的关键正向调控因子,主要作用于自噬的起始阶段。
从机制层面来看,TRIM49与自噬起始核心蛋白ULK1相互作用。当TRIM49缺陷时,会下调ULK1的蛋白表达水平,从而抑制自噬的启动。而患者来源的突变则会破坏TRIM49对自噬的正向调控能力。研究还发现TRIM49的表达受miR-548b-3p和视网膜发育关键转录因子OTX2的调控,三者形成了一个复杂的调控网络,共同维持RPE细胞的正常功能。
该研究基于眼遗传病家系样本资源库及高通量测序技术,通过细胞模型及分子机制研究,首次将TRIM49确立为常染色体隐性遗传RP的新致病基因,并将自噬、吞噬与RP发病机制紧密联系起来。其研究结果为理解感光细胞变性疾病的病理过程提供了全新视角。
相关论文信息:https://doi.org/10.1002/advs.202512305
版权声明:凡本网注明“来源:中国科学报、科学网、科学新闻杂志”的所有作品,网站转载,请在正文上方注明来源和作者,且不得对内容作实质性改动;微信公众号、头条号等新媒体平台,转载请联系授权。邮箱:shouquan@stimes.cn。






